\(\renewcommand{\AA}{\text{Å}}\)
improper_style distance command
Syntax
improper_style distance
Examples
improper_style distance
improper_coeff 1 80.0 100.0
Description
The distance improper style uses the potential
where \(d\) is the distance between the central atom and the plane formed by the other three atoms. If the 4 atoms in an improper quadruplet (listed in the data file read by the read_data command) are ordered I,J,K,L then the I-atom is assumed to be the central atom.
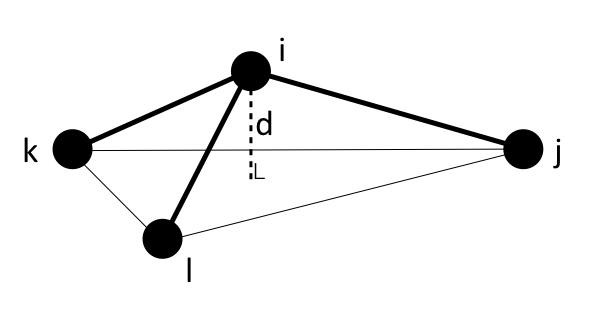
Note that defining 4 atoms to interact in this way, does not mean that bonds necessarily exist between I-J, J-K, or K-L, as they would in a linear dihedral. Normally, the bonds I-J, I-K, I-L would exist for an improper to be defined between the 4 atoms.
The following coefficients must be defined for each improper type via the improper_coeff command as in the example above, or in the data file or restart files read by the read_data or read_restart commands:
\(K_2\) (energy/distance^2)
\(K_4\) (energy/distance^4)
Restrictions
This improper style can only be used if LAMMPS was built with the MOLECULE package. See the Build package doc page for more info.
Default
none